HPLC分析方法開發(fā)與驗證系統(tǒng)全面講解
一���、分析方法開發(fā)
分析方法的開發(fā)主要包括色譜柱的選擇、流動相的選擇����、檢測波長的選擇和梯度的優(yōu)化幾個方面����。目前高效液相多做反相使用,所以本文主要以反相為例進(jìn)行講解。
1.色譜柱的選擇
原料藥生產(chǎn)對產(chǎn)品的純度和雜質(zhì)含量的要求非?����?量?,要求檢測使用的色譜柱有較高的理論塔板數(shù),能提供更好的分離度�����,從而對可能存在的雜質(zhì)有更大的分離的可能性�,所以5um填料的色譜柱長要250mm,3.5um填料的柱長要150mm����,基本上都是各個粒徑柱長最長的。我比較喜歡近兩年新出的亞二微米填料的色譜柱�,50mm柱長就能提供很高的理論塔板數(shù),而且柱長和粒徑小了�����,流速增加很多�����,能節(jié)省很多的分析時間,極大的提高工作效率�����。一般選用直徑為4.6mm或3.0mm的柱子��,太細(xì)了可能會增大柱外效應(yīng)�。填料的孔徑對于小分子合成藥物不需要考慮,普通的分析柱都在100A左右�����,能滿足分析檢測的需要���。
對于API分析方法開發(fā)��,一般要求必須做色譜柱的篩選實驗�����,最少使用三種不同類型的色譜柱,每種類型三只����,要來自于不同廠家�����。
三種類型包括:
1)普通的C18或相應(yīng)的C8色譜柱����,如Waters的Symmetry C18或C8��,YMC的Pack Pro C18或C8���,Agilent的RX C8等��,其它公司如菲羅門和熱電也有相應(yīng)的色譜柱�����;
2)封端處理的或者極性嵌入型色譜柱�����,如Waters的SymmetryShield RP18或RP8���,XTerra RP18或RP8,YMC的ODS AQ��,Agilent的Zorbax SB AQ等,其它公司如菲羅門和熱電也有相應(yīng)的色譜柱����;
3)填料用其它官能團(tuán)修飾過的色譜柱,如苯基柱等��,很多公司都有�。
一般不同類型的色譜柱在選擇性上會有很大的差異,相同類型的色譜柱生產(chǎn)廠家不同在選擇性上也會有差異�,這個主要是填料的性質(zhì)和生產(chǎn)工藝決定的,有時候用一只色譜柱分離不好���,除了優(yōu)化梯度和流動相外����,換一個廠家的柱子也是一個很好的選擇�。相同品牌型號的色譜柱,C18和C8在選擇性上沒有差異����,但是C18保留能力更強,相同的樣品分離度更高����,我們一般傾向于選擇用C18。我們在篩選色譜柱時盡量選擇行業(yè)內(nèi)排名前幾位的廠家���,柱子品質(zhì)好�,開發(fā)分析方法時能省很多力氣����,做出來的分析方法也有保證。一個藥從開發(fā)到上市可能會持續(xù)十幾年甚至更長時間����,廠家有實力,開發(fā)方法時選定的柱子在若干年以后需要時還會有的買�,做分析時重復(fù)性也能保障。多用幾只色譜柱做篩選和分析方法優(yōu)化�����,能盡最大的可能提高分析方法的質(zhì)量�����,保證檢測結(jié)果的可信度�����。
我比較喜歡用的柱子有:Agilent的Zorbax EclipseXDB-C18、ZorbaxEclipse Plus C18��,Waters的Symmetry C18�����、XTerra RP18�����、XTerra MS C18等�����,YMC的柱子有時會是不錯的備用選擇�,菲羅門的柱子菲羅門的柱子國內(nèi)外市場占有率較高,但是感覺柱子壓力高�����,壽命短��,盡量不用����,熱電的柱子用的也不少�,但沒什么感覺����。
制劑分析方法選擇色譜柱的要求基本和API相一樣�����,對于中間體和生產(chǎn)過程中反應(yīng)跟蹤(IPC)分析方法開發(fā)使用的色譜柱��,我們一般會根據(jù)樣品性質(zhì)直接選取一兩只普通C18或者封端處理過的色譜柱����,簡化篩選過程。
2.流動相的選擇
常用做反相流動相的溶劑是甲醇和乙腈����,甲醇有其性價比的優(yōu)勢,但是甲醇活性高�����,可能與某些樣品發(fā)生反應(yīng)�����,而且甲醇在低波長下有紫外吸收,會降低分析方法的靈敏度�;乙腈雖然價格很高,毒性比甲醇大���,但是洗脫能力比甲醇強�����,很少與樣品發(fā)生反應(yīng)���,用作流動相系統(tǒng)壓力要比甲醇低很多,且截止波長比甲醇低20nm����,增加了檢測出在低波長下才有吸收的雜質(zhì)的可能性,所以我們一般傾向于多用乙腈�����,少用甲醇�����。但是有時候樣品峰形不好或者分離不好,更換溶劑試試是一個很好的選擇�,畢竟不同的溶劑提供不同的選擇性。
對流動相的優(yōu)化主要在水相上下功夫�,水里可以加酸、加堿����、加鹽,從而改善峰形���、提高分離度。流動相里加堿的情況比較少����,主要還是加酸,常用的酸有磷酸���、三氟乙酸�、甲酸�、乙酸、高氯酸���、甲基磺酸等�����,其中最常用的是磷酸和三氟乙酸�����,磷酸在低波長下沒有紫外吸收�����,而三氟乙酸在低波長下有����,但是三氟乙酸易揮發(fā)而磷酸不行,所以單純做液相�����,低波長下磷酸最合適����,三氟乙酸有吸收,運行梯度時基線漂移很嚴(yán)重��,而做液質(zhì)就要考慮首選三氟乙酸了�����,近些年還比較流行加甲酸或乙酸。一般情況下這幾種酸沒有太大區(qū)別�����,我們更多的是考慮通過加酸改變流動相的pH值���,從而改善樣品的分離度和峰形��。下面兩圖是流動相的pH值改變對一組樣品分離和峰形的影響�����。

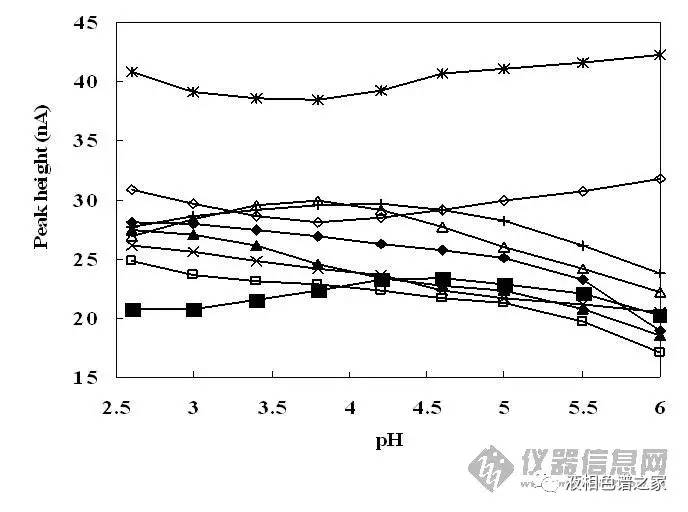
從圖中可以看出:
1)流動相pH值改變可以改變樣品的保留時間和分離度;
2)流動相pH值改變甚至可以改變某些樣品出峰的先后順序����;
3)流動相pH值改變可以改變樣品的峰高,即可以調(diào)整峰形����。
相同進(jìn)樣量樣品峰越高則意味著峰形越好,從圖中可以看出多數(shù)樣品在低pH值下峰形都比中性要好�,這個主要是由色譜柱本身的性質(zhì)所決定的�。色譜柱主要都是硅膠基質(zhì)���,現(xiàn)有的填料處理工藝無法將硅膠上殘余的硅羥基全部去除���,硅羥基會造成樣品峰拖尾,一般認(rèn)為硅羥基的pKa在3.5到4.5之間�,低pH值能幫助抑制硅羥基的活性,減小拖尾����,從而改善峰形,提高分離度�����。水溶液中添加0.1%(體積)的磷酸或者三氟乙酸其pH值大概在2左右����,用作流動相正好抑制硅羥基的活性,所以開發(fā)液相分析方法時流動相首選水加01.%的磷酸�,然后再以此為基礎(chǔ)做優(yōu)化。
在單獨用酸不行的時候就要考慮使用緩沖鹽����,緩沖鹽的選擇原則是:簡單���、穩(wěn)定、緩沖能力強�����、配制簡單���,需要調(diào)pH值時要有相應(yīng)的酸或堿�����。常用的緩沖鹽是磷酸鹽����,主要是鉀鹽和鈉鹽��,再有就是醋酸鹽��,常用的鹽濃度在10~20mM左右�。以前因為色譜柱填料生產(chǎn)工藝的問題�����,往往需要在流動相里添加三乙胺來減少拖尾,但是三乙胺對色譜柱的壽命有很大影響�,現(xiàn)在新的色譜柱都不再需要了。流動相里有時會需要調(diào)節(jié)pH值到堿性���,具體pH要視色譜柱的耐受范圍而定�����,常用NaOH����、KOH溶液或氨水做為調(diào)節(jié)緩沖鹽溶液堿性pH的試劑�,也可以往水里單獨添加氨水做堿性流動相。
在緩沖鹽做流動相時�����,出峰太早����、峰形很差、相似結(jié)構(gòu)的化合物峰因為拖尾或峰型太寬而不能達(dá)到基線分離時���,可以考慮使用離子對試劑����,常用的離子對試劑主要是各種烷基磺酸鈉和四丁基銨鹽,但是流動相里使用離子對試劑時����,系統(tǒng)需要的平衡時間長,樣品保留時間不是很穩(wěn)定�,因為離子對試劑的背景吸收基線會很差,且做完樣品后需要長時間清洗��,所以我們盡量不使用離子對試劑��。
使用緩沖鹽時要注意流動相混合以后鹽可能析出的問題和鹽背景吸收導(dǎo)致基線漂移嚴(yán)重的問題�����,尤其是在流動相里添加醋酸銨以后�����,在低波長下梯度變化時基線下降非常嚴(yán)重��,嚴(yán)重影響對含量較小雜質(zhì)的準(zhǔn)確定量����,可以考慮在乙腈里加入10%的水,水中預(yù)先加入10倍水溶液濃度的緩沖鹽����,這樣梯度中A、B兩項鹽的濃度相同����,可以避免基線漂移嚴(yán)重的問題。
原料藥一般結(jié)構(gòu)式比較大�,分子構(gòu)成比較復(fù)雜,開發(fā)分析方法時用水加磷酸效果可能效果不好���,通常還要求最少嘗試2和6.5兩個pH值的磷酸鹽緩沖溶液�,并依據(jù)結(jié)果對流動相進(jìn)行pH優(yōu)化���,如效果不理想再進(jìn)一步嘗試其它緩沖鹽溶液�。開發(fā)中間體或者IPC的分析方法時可以根據(jù)經(jīng)驗酌情簡化流動相的選擇過程���。
3.梯度的優(yōu)化
梯度優(yōu)化主要是通過調(diào)節(jié)流動相的起始比例和梯度的斜率來調(diào)整樣品的保留時間����,優(yōu)化樣品的分離度。下圖是梯度中有機相起始比例的變化對一組樣品分離的影響�����。
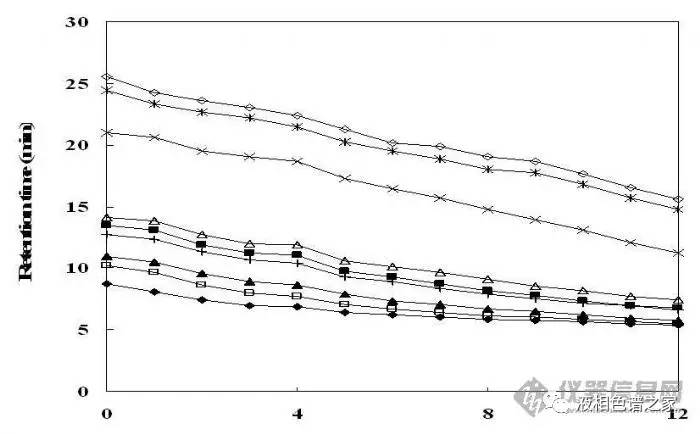
從圖中可以看出:有機相起始比例越小��,樣品保留時間越長�,隨著梯度的改變,樣品出峰的先后順序也有可能改變�。我們做梯度優(yōu)化時主要調(diào)整梯度的起始比例和斜率。現(xiàn)在的色譜柱或者采用了新的封端工藝��,或者內(nèi)嵌極性基團(tuán)�����,耐水的能力都比較高����。在酸性條件下很多化合物都以離子形式存在,極性較大�����,為了提高樣品的分離度����,盡量使用大比例的水做梯度的起始����。對于添加緩沖鹽的流動相要注意梯度變化過程中流動相組成改變時不能有鹽析出���。
對于水加0.1%磷酸的流動相,開始時可以采用95%的水做起始����,以95%的有機相結(jié)束,注意根據(jù)實際情況在梯度最后用大比例的有機相沖洗幾分鐘�����,以保證把小極性的雜質(zhì)洗脫下來��,防止樣品殘留到下一針�����。梯度的斜率一般采用凹線型的先小后大�����,梯度變化先慢后快,在此基礎(chǔ)上再對梯度進(jìn)行優(yōu)化�����。
使用緩沖鹽溶液的梯度水相起始比例一般要從10~20%開始����,為了防止鹽析出,在梯度最后避免用純的有機相做沖洗�����,梯度斜率采用恒定的就可以����,在此基礎(chǔ)上根據(jù)方法運行的情況對梯度進(jìn)行調(diào)整。
一般一個API的樣品采集的時間控制在40~50分鐘左右���,樣品出峰在15~20分鐘左右比較好��,如果有極性非常小的雜質(zhì)存在可以在最后加一段時間的大比例有機溶劑沖洗色譜柱��,最后再設(shè)置10分鐘左右的重新平衡時間�����。中間體和IPC的樣品分析方法時間可以根據(jù)需要減半或者時間更短�。
4.波長的選擇
做分析方法開發(fā)需要二極管陣列檢測器,做色譜峰純度檢查和選擇檢測波長�����,通過色譜峰純度檢查來保證主峰里沒有掩蓋其它雜質(zhì)���,做純度檢測對波長選擇的要求比較簡單,原則是把盡量多的雜質(zhì)在色譜圖上體現(xiàn)出來�����。很多雜質(zhì)只有在低波長下才有紫外吸收�����,所以我們選擇盡可能低的波長�,乙腈的截止波長在190~195nm,用乙腈做流動相檢測波長可以選擇在210~220nm�。也有公司要求分別選取產(chǎn)品紫外吸收最強的波段和210~220nm兩個波段做對比,哪個純度低以哪個為準(zhǔn)�,這樣可以更嚴(yán)格的控制產(chǎn)品的質(zhì)量。
二�����、分析方法驗證
為了保證分析檢測結(jié)果準(zhǔn)確、可靠�,必須對所采用的分析方法的準(zhǔn)確性、科學(xué)性和可行性進(jìn)行驗證���,以證明分析方法符合檢測的目的和要求����,這就是分析方法驗證���。從本質(zhì)上講�����,方法驗證就是根據(jù)檢測項目的要求����,預(yù)先設(shè)置一定的驗證內(nèi)容���,并通過設(shè)計合理的試驗來驗證所采用的分析方法符合檢測項目的要求���。方法驗證在質(zhì)量控制上有重要的作用和意義��,只有經(jīng)過驗證的分析方法才能用于藥品生產(chǎn)的分析檢測�,方法驗證是制訂質(zhì)量標(biāo)準(zhǔn)的基礎(chǔ)�。方法驗證內(nèi)容包括方法的專屬性、線性���、范圍�����、準(zhǔn)確度、精密度�、檢出限、定量限����、耐用性和系統(tǒng)適用性等,檢測目的不同驗證要求也不盡相同�。
1.專屬性
專屬性是指分析方法能夠?qū)a(chǎn)品和雜質(zhì)分開的特性,也稱為選擇性�。對于純度檢測,可在標(biāo)準(zhǔn)品中加入產(chǎn)品中的已知雜質(zhì)����,或者直接用粗品�,考察產(chǎn)品峰是否受到雜質(zhì)的干擾����,對于過程跟蹤,可用反應(yīng)體系樣品來考察有沒有其它的雜質(zhì)干擾�����。必要時使用二極管陣列檢測器或者質(zhì)譜檢測器進(jìn)行色譜峰純度檢查�。一般要求產(chǎn)品和雜質(zhì)之間的分離度大于2.0。
2.線性
線性是在設(shè)定的范圍內(nèi)��,檢測結(jié)果與樣品中原料或產(chǎn)品的濃度呈線性關(guān)系的程度�����。線性是定量檢測的基礎(chǔ)���,需要定量檢測的項目都需要驗證線性��。一般用貯備液經(jīng)過精密稀釋��,或分別精密稱樣����,制備得到一系列被測物質(zhì)的濃度(5個以上),按濃度從小到大運行序列���,以峰面積和濃度的函數(shù)作圖���,用最小二乘法進(jìn)行線性回歸計算,考察分析方法的線性���。
3.范圍
范圍指在能夠達(dá)到一定的準(zhǔn)確度�、精密度和線性時�,樣品中被分析物的濃度區(qū)間。簡單的說���,范圍就是分析方法適用的樣品中待測物的濃度最大值和最小值。需要定量檢測的分析方法都需要對范圍進(jìn)行驗證��,純度檢測時����,范圍應(yīng)為測試濃度的80%~120%。
4.準(zhǔn)確度
準(zhǔn)確度是指測定的結(jié)果與真實值之間接近的程度�,所以也叫做真實度,需要定量得分析方法均需要驗證準(zhǔn)確度。準(zhǔn)確度應(yīng)在規(guī)定的范圍內(nèi)建立�����,對于原料藥可用已知純度的標(biāo)準(zhǔn)品或符合要求的原料藥進(jìn)行測定��,必要時可與另一個已建立準(zhǔn)確度的方法比較結(jié)果����。
5.精密度
精密度是指在規(guī)定條件下,同一均勻樣品經(jīng)多次取樣進(jìn)行一系列檢測所得結(jié)果之間的接近程度��。精密度一般用相對標(biāo)準(zhǔn)偏差表示���,取樣檢測次數(shù)應(yīng)至少6次�。
精密度可以從三個層次考察:重復(fù)性�����、中間精密度��、重現(xiàn)性��。
a�、重復(fù)性是在相同的操作條件下�、較短時間間隔內(nèi)�����,由同一分析人員測定所得結(jié)果的精密度���。一般是用100%濃度水平的樣品測定6次的結(jié)果進(jìn)行評價����。
b��、中間精密度:同一實驗室�,在日期、分析人員����、儀器等內(nèi)部條件改變時,測定結(jié)果的精密度�����。
c���、重現(xiàn)性:指不同實驗室之間不同分析人員測定結(jié)果的精密度。
6.檢出限
檢出限是指樣品中的被分析物能夠被檢測到的最低量,不需要準(zhǔn)確定量���。檢出限體現(xiàn)了分析方法的靈敏度��。檢出限的測定可以通過對一系列已知濃度被測物的試樣進(jìn)行檢測���,以能準(zhǔn)確、可靠檢出被測物的最小濃度來確定�����,也可把已知濃度樣品的信號與噪聲信號進(jìn)行比較�����,以信噪比為3:1時的濃度確定檢出限���,一般要求能夠達(dá)到進(jìn)樣濃度的0.05%��。
7.定量限
定量限是指樣品中的被分析物能夠被定量檢測的最低量����,其測定結(jié)果需要一定的準(zhǔn)確度和精密度��,定量限體現(xiàn)了分析方法靈敏定量檢測的能力。檢測需要嚴(yán)格控制含量的雜質(zhì)�,必須考察方法的定量限,以保證雜質(zhì)能夠被準(zhǔn)確定量����。一般以信噪比為10:1時相應(yīng)的濃度或進(jìn)樣量來確定定量限。
8.耐用性
耐用性是指測定條件發(fā)生小的變動時���,測定結(jié)果不受影響的承受程度��,耐用性主要表明方法的抗干擾能力����,主要的變動因素包括:流動相的組成��、流速和pH值��、色譜柱���、柱溫等�。經(jīng)試驗����,應(yīng)說明小的變動能否符合系統(tǒng)適用性試驗要求,以確保方法有效��。
9.系統(tǒng)適用性試驗
液相色譜分析方法主要依賴高效液相色譜儀和色譜柱�,在做方法驗證時,有必要將高效液相色譜儀����、色譜柱、流動相與實驗操作�、待測樣品等一起當(dāng)作完整的系統(tǒng)進(jìn)行評估,并將系統(tǒng)適用性作為分析方法的組成部分���,系統(tǒng)適用性便是對整個系統(tǒng)進(jìn)行評估的指標(biāo)�����。一般系統(tǒng)適應(yīng)性的要求為:分析方法能夠達(dá)到0.05%的檢出限�,主峰的拖尾因子0.5<Tf<2.5���,主峰與雜質(zhì)的分離度大于2.0���,空白干凈,主峰處無系統(tǒng)峰干擾�。
三��、總結(jié)
分析方法開發(fā)與驗證是一個整體�����,在實際工作中��,一般是先開發(fā)分析方法�,經(jīng)過適當(dāng)?shù)膬?yōu)化以后再做方法驗證���,驗證的部分內(nèi)容在分析方法開發(fā)時就要做�,比如說分析方法的專屬性驗證�。分析方法驗證并非必須驗證所有的內(nèi)容,只要注意驗證內(nèi)容充分�,足以證明分析方法的合理性就可以了,如雜質(zhì)限度檢測一般只需要驗證專屬性和檢出限��,而精密度��、線性�����、定量限等涉及定量測定的項目,則不需要驗證�����。
有時需要對分析方法進(jìn)行全面或部分的再驗證�。當(dāng)原料藥合成工藝發(fā)生改變時����,可能引入新的雜質(zhì),雜質(zhì)檢查方法和含量測定方法的專屬性就需要再進(jìn)行驗證����,以證明有關(guān)物質(zhì)檢查方法能夠檢測新引入的雜質(zhì),且新引入的雜質(zhì)對主成份的含量測定應(yīng)無干擾���。當(dāng)分析方法發(fā)生部分改變時�,如檢測波長發(fā)生改變�����,則需要重新進(jìn)行檢測限�����、專屬性��、準(zhǔn)確度、精密度���、線性等內(nèi)容的驗證��,以證明改變后的分析方法的合理性�、可行性���。


